We're for all- ALL are for us for the greater interest of Humanism-Truth-Facts-Friendship-Unity-Participation including Physico-Mental Sound Health with Spirituality, enrichment through ''TOTAL HEALTH SOLUTION'' to a Well-furnished GOALofTruth alloted for all in real sense ;
From wikipedia & other reliable sources ( Poets, Writers, Thinkers, Researchers, Free Lancers, Philosophers, Theologists, Scientists, Orators, Sociologists and Photographers +Artists-Musicians & etc.) we can learn as follows :
![]()

''Thalassemias are inherited blood disorders characterized by decreased hemoglobin production.[7] Symptoms depend on the type and can vary from none to severe.[1] Often there is mild to severe anemia (low red blood cells or hemoglobin).[1] Anemia can result in feeling tired and pale skin.[1] There may also be bone problems, an enlarged spleen, yellowish skin, and dark urine.[1] Slow growth may occur in children.[1]
Thalassemias are genetic disorders inherited from a person's parents.[2] There are two main types, alpha thalassemia and beta thalassemia.[7] The severity of alpha and beta thalassemia depends on how many of the four genes for alpha globin or two genes for beta globin are missing.[2] Diagnosis is typically by blood tests including a complete blood count, special hemoglobin tests, and genetic tests.[3] Diagnosis may occur before birth through prenatal testing.[8]
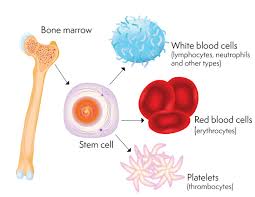
Treatment depends on the type and severity.[4] Treatment for those with more severe disease often includes regular blood transfusions, iron chelation, and folic acid.[4] Iron chelation may be done with deferoxamine, deferasirox or deferiprone.[4][9] Occasionally, a bone marrow transplant may be an option.[4] Complications may include iron overload from the transfusions with resulting heart or liver disease, infections, and osteoporosis.[1] If the spleen becomes overly enlarged, surgical removal may be required.[1] Thalassemia patients who do not respond well to blood transfusions can take hydroxyurea or thalidomide, and sometimes a combination of both.[10] Hydroxyurea is the only FDA approved drug for thalassemia. Patients who took 10 mg/kg of hydroxyurea every day for a year had significantly higher hemoglobin levels, and it was a well-tolerated treatment for patients who did not respond well to blood transfusions.[11] Another hemoglobin-inducer includes thalidomide, although it has not been tested in a clinical setting. The combination of thalidomide and hydroxyurea resulted in hemoglobin levels increasing significantly in transfusion-dependent and non-transfusion dependent patients [12]
As of 2015, thalassemia occurs in about 280 million people, with about 439,000 having severe disease.[13] It is most common among people of Greek, Italian, Middle Eastern, South Asian, and African descent.[7] Males and females have similar rates of disease.[14] It resulted in 16,800 deaths in 2015, down from 36,000 deaths in 1990.[6][15] Those who have minor degrees of thalassemia, similar to those with sickle-cell trait, have some protection against malaria, explaining why they are more common in regions of the world where malaria exists''.[16]
''Signs and symptoms[edit]
 Left: Hand of a person with severe anemia. Right: Hand of a person without anemia.
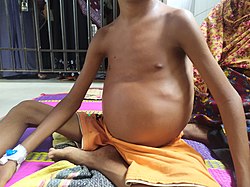
Left: Hand of a person with severe anemia. Right: Hand of a person without anemia. A patient having thalassemia shows enlarged spleen.
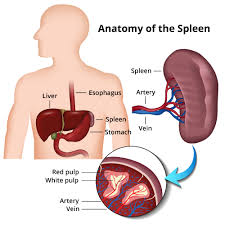
A patient having thalassemia shows enlarged spleen.- Iron overload: People with thalassemia can get an overload of iron in their bodies, either from the disease itself or from frequent blood transfusions. Too much iron can result in damage to the heart, liver, and endocrine system, which includes glands that produce hormones that regulate processes throughout the body. The damage is characterized by excessive deposits of iron. Without adequate iron chelation therapy, almost all patients with beta-thalassemia accumulate potentially fatal iron levels.[17]
- Infection: People with thalassemia have an increased risk of infection. This is especially true if the spleen has been removed.[18]
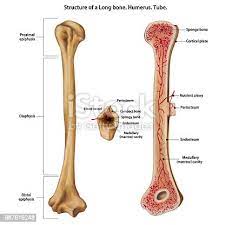
- Bone deformities: Thalassemia can make the bone marrow expand, which causes bones to widen. This can result in abnormal bone structure, especially in the face and skull. Bone marrow expansion also makes bones thin and brittle, increasing the risk of broken bones.[19]
- Enlarged spleen: The spleen aids in fighting infection and filters unwanted material, such as old or damaged blood cells. Thalassemia is often accompanied by the destruction of a large number of red blood cells and the task of removing these cells causes the spleen to enlarge. Splenomegaly can make anemia worse, and it can reduce the life of transfused red blood cells. Severe enlargement of the spleen may necessitate its removal.[20]
- Slowed growth rates: anemia can cause the growth of a child to slow down. Puberty may also be delayed in children with thalassemia.[21]
- Heart problems: Diseases, such as congestive heart failure and abnormal heart rhythms, may be associated with severe thalassemia.[22]
Hemoglobin structural biology[edit]
Normal human hemoglobins are tetrameric proteins composed of two pairs of globin chains, each of which contains one alpha-like (α-like) chain and one beta-like (β-like) chain. Each globin chain is associated with an iron-containing heme moiety. Throughout life, the synthesis of the alpha-like and the beta-like (also called non-alpha-like) chains is balanced so that their ratio is relatively constant and there is no excess of either type.[23]
The specific alpha and beta-like chains that are incorporated into Hb are highly regulated during development:
- Embryonic Hbs are expressed as early as four to six weeks of embryogenesis and disappear around the eighth week of gestation as they are replaced by fetal Hb.[24][25] Embryonic Hbs include:
- Hb Gower-1, composed of two ζ globins (zeta globins) and two ε globins (epsilon globins) (ζ2ε2)
- Hb Gower-2, composed of two alpha globins and two epsilon globins (α2ε2)
- Hb Portland, composed of two zeta globins and two gamma globins (ζ2γ2)
- Fetal Hb (Hb F) is produced from approximately eight weeks of gestation through birth and constitutes approximately 80 percent of Hb in the full-term neonate. It declines during the first few months of life and, in the normal state, constitutes <1 percent of total Hb by early childhood. Hb F is composed of two alpha globins and two gamma globins (α2γ2).
- Adult Hb (Hb A) is the predominant Hb in children by six months of age and onward; it constitutes 96-97% of total Hb in individuals without a hemoglobinopathy. It is composed of two alpha globins and two beta globins (α2β2).[citation needed]
- Hb A2 is a minor adult Hb that normally accounts for approximately 2.5-3.5% of total Hb from six months of age onward. It is composed of two alpha globins and two delta globins (α2δ2).[citation needed]
Cause[edit] Thalassemia has an autosomal recessive pattern of inheritance.
Both α- and β-thalassemias are often inherited in an autosomal recessive manner. Cases of dominantly inherited α- and β-thalassemias have been reported, the first of which was in an Irish family with two deletions of 4 and 11 bp in exon 3 interrupted by an insertion of 5 bp in the β-globin gene. For the autosomal recessive forms of the disease, both parents must be carriers for a child to be affected. If both parents carry a hemoglobinopathy trait, the risk is 25% for each pregnancy for an affected child.[citation needed]
The genes involved in thalassemia control the production of healthy hemoglobin. Hemoglobin binds oxygen in the lungs and releases it when the red cells reach peripheral tissues, such as the liver. The binding and release of oxygen by hemoglobin are essential for survival.[citation needed]
Evolution[edit]
Having a single genetic variant for thalassemia may protect against malaria and thus can be an advantage.[26]
People diagnosed with heterozygous (carrier) β-thalassemia have some protection against coronary heart disease.[27]
Pathophysiology[edit]
Normally, the majority of adult hemoglobin (HbA) is composed of four protein chains, two α and two β-globin chains arranged into a heterotetramer. In thalassemia, patients have defects in either the α or β-globin chain, causing production of abnormal red blood cells.[citation needed]
The thalassemias are classified according to which chain of the hemoglobin molecule is affected. In α-thalassemias, production of the α-globin chain is affected, while in β-thalassemia, production of the β-globin chain is affected.[28]
The β-globin chains are encoded by a single gene on chromosome 11; α-globin chains are encoded by two closely linked genes on chromosome 16.[29] Thus, in a normal person with two copies of each chromosome, two loci encode the β chain, and four loci encode the α chain. Deletion of one of the α loci has a high prevalence in people of African or Asian descent, making them more likely to develop α-thalassemia. β-Thalassemias are not only common in Africans, but also in Greeks and Turks.[citation needed]
Alpha-thalassemias[edit]
The α-thalassemias involve the genes HBA1[30] and HBA2,[31] inherited in a Mendelian recessive fashion. Two gene loci and so four alleles exist. Two genetic loci exist for α-globin, thus four alleles are in diploid cells. Two alleles are maternal and two alleles are paternal in origin. The severity of the α-thalassemias is correlated with the number of affected α-globin; alleles: the greater, the more severe will be the manifestations of the disease.[32] Alpha-thalassemias result in decreased alpha-globin production; therefore, fewer alpha-globin chains are produced, resulting in an excess of β chains in adults and excess γ chains in newborns. The excess β chains form unstable tetramers (called hemoglobin H or HbH of 4 beta chains), which have abnormal oxygen dissociation curves. Alpha thalassemias often are found in people from Southeast Asia, the Middle East, China, and in those of African descent.[33]
| # of missing alleles | Types of alpha thalassemia[32] | Symptoms |
|---|---|---|
| 1 | Silent carrier | No symptoms |
| 2 | Alpha thalassemia trait | Minor anemia |
| 3 | Hemoglobin H disease | Mild to moderate anemia; may lead normal life |
| 4 | Hydrops fetalis | Death usually occurs in utero or at birth |
Beta-thalassemia[edit]
Beta thalassemias are due to mutations in the HBB gene on chromosome 11,[34] also inherited in an autosomal, recessive fashion. The severity of the disease depends on the nature of the mutation and on the presence of mutations in one or both alleles.
Mutated alleles are called β+ when partial function is conserved (either the protein has a reduced function, or it functions normally but is produced in reduced quantity) or βo, when no functioning protein is produced.
The situation of both alleles determines the clinical picture:
- β thalassemia major (Mediterranean anemia or Cooley anemia) is caused by a βo/βo genotype. No functional β chains are produced, and thus no hemoglobin A can be assembled. This is the most severe form of β-thalassemia;
- β thalassemia intermedia is caused by a β+/βo or β+/β+ genotype. In this form, some hemoglobin A is produced;
- β thalassemia minor is caused by a β/βo or β/β+ genotype. Only one of the two β globin alleles contains a mutation, so β chain production is not terribly compromised and patients may be relatively asymptomatic.
Beta thalassemia most often occurs in people of Mediterranean origin. To a lesser extent, Chinese, other Asians, and African Americans can be affected.[33]
Delta-thalassemia[edit]
As well as alpha and beta chains present in hemoglobin, about 3% of adult hemoglobin is made of alpha and delta chains. Just as with beta thalassemia, mutations that affect the ability of this gene to produce delta chains can occur.[35][36]
Combination hemoglobinopathies[edit]
Thalassemia can coexist with other hemoglobinopathies. The most common of these are:
- Hemoglobin E/thalassemia: common in Cambodia, Thailand, and parts of India, it is clinically similar to β thalassemia major or thalassemia intermedia.[citation needed]
- Hemoglobin S/thalassemia: common in African and Mediterranean populations, it is clinically similar to sickle-cell anemia, with the additional feature of splenomegaly.[citation needed]
- Hemoglobin C/thalassemia: common in Mediterranean and African populations, hemoglobin C/βo thalassemia causes a moderately severe hemolytic anemia with splenomegaly; hemoglobin C/β+ thalassemia produces a milder disease.[citation needed]
- Hemoglobin D/thalassemia: common in the northwestern parts of India and Pakistan (Punjab region).[37]
Diagnosis[edit]
Thalassemia can be diagnosed via a complete blood count, hemoglobin electrophoresis or high-performance liquid chromatography, and DNA testing.[38][39] Hemoglobin electrophoresis is not widely available in developing countries, but the Mentzer index can also be used for diagnosis of thalassemia; it is not a definitive test but it can suggest the possibility of thalassemia. The Mentzer index can be calculated from a complete blood count report.[40]
Prevention[edit]
The American College of Obstetricians and Gynecologists recommends all people thinking of becoming pregnant be tested to see if they have thalassemia.[41] Genetic counseling and genetic testing are recommended for families who carry a thalassemia trait.[citation needed]
A screening policy exists in Cyprus to reduce the rate of thalassemia, which, since the program's implementation in the 1970s (also including prenatal screening and abortion), has reduced the number of children born with the disease from one of every 158 births to almost zero.[42] Greece also has a screening program to identify people who are carriers.[43]
In Iran as a premarital screening, the man's red cell indices are checked first. If he has microcytosis (mean cell hemoglobin < 27 pg or mean red cell volume < 80 fl), the woman is tested. When both are microcytic, their hemoglobin A2 concentrations are measured. If both have a concentration above 3.5% (diagnostic of thalassemia trait) they are referred to the local designated health post for genetic counseling.[44]
Large-scale awareness campaigns are being organized in India[45] both by government and non-government organizations to promote voluntary premarital screening, with marriage between carriers strongly discouraged''.
We're for all- ALL are for us for the greater interest of Humanism-Truth-Facts-Friendship-Unity-Participation to a Well-furnished GOAL of Truth from which all shall have ++++;
We're indebted to WIKIPEDIA +WHO for a short while and as 'Guardian QUOTATION' from Global WISER ONE. And have quoted many images, article's, writings etc. by great & humanist writers+++ from global thinkers, Well-wishers, Wiseman, Humanists and Others Living-Nonlivings in favor of HUMANISM to share more answers of Researchers-readers+++++....
To reach the 'GOAL of FULFILNESS' unitedly to alive in the "DESTINATION of TRUTH-FACTS-CHARMEST AMICABLITY" of Natural Joyful POSSIBILITIES+++
BREAST CANCER, TUMOR, Arsenic+Chemical Poisoning, Corona, Heart-Lung diseases, Neurological-Hormonal-Immunal-Infectious diseases with related complexities are possible to cure properly-easily-scientifically-accurately (100%) by our positive medical services only with+++++balance confirming
Medicine-Food control-proper nursing-medicinal massage-Medicinal Yogas- Meditation, Physiotherapy special etc. without side effects & Repeatation as per contract through user-friendly approved ways of CURE++++. please fill our form as below or click:
After confirming contract-letter between you+++. We serve you properly with no Chemo-therapy-radiation therapy -SURGICAL Complexities (Physical-Mental) to CURE+++ upto our Limit to recover your both-health from illness.